
Brookhaven Lab chemist Ping Liu and Stony Brook University graduate student Hong Zhang developed a theoretical framework to predict the behavior of catalysts under reaction conditions. The framework identifies how reaction conditions can then be used to tune both catalytic performance and selectivity.
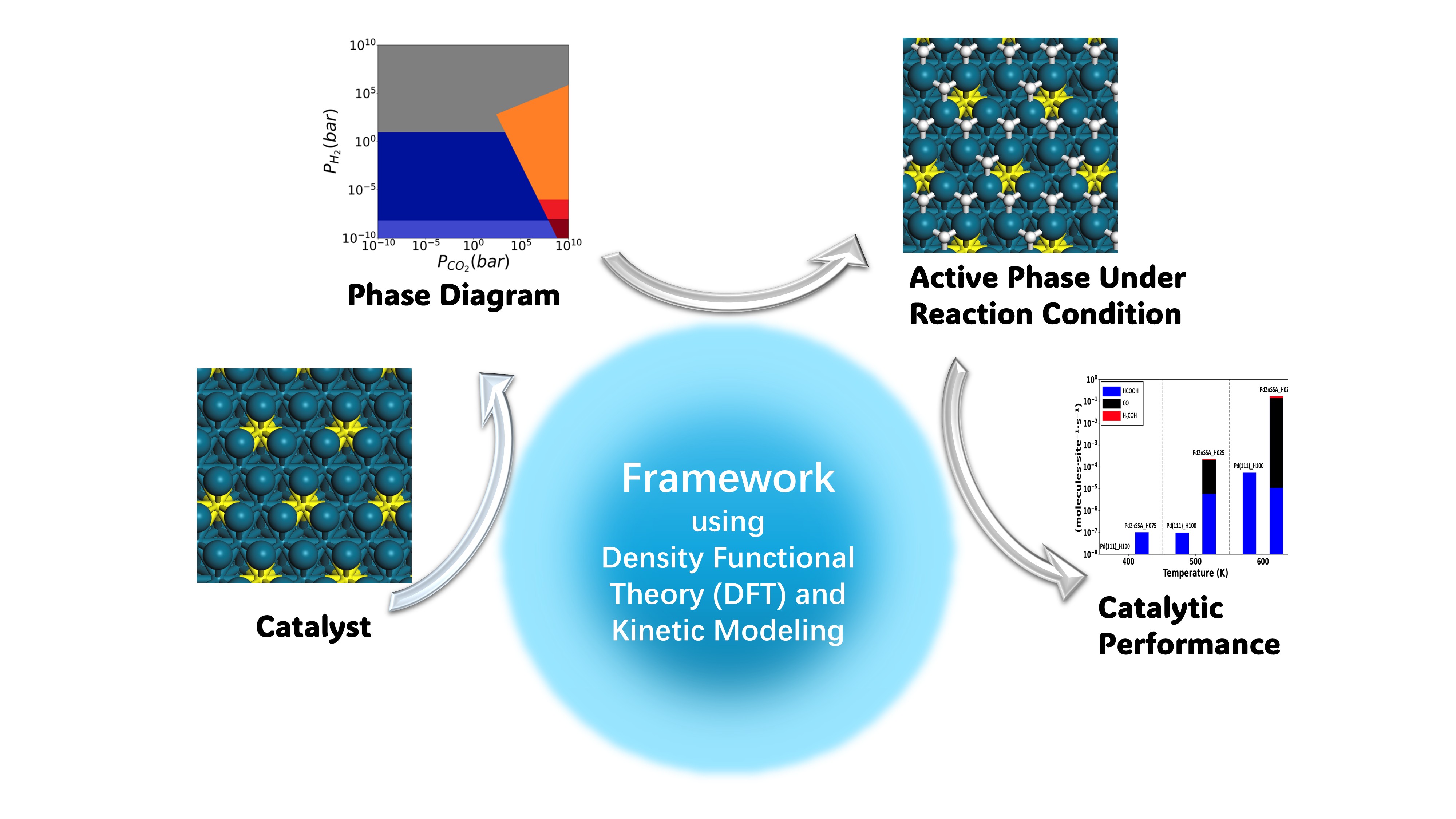
Schematic of the theoretical framework: Starting with a computational model of the prepared catalyst, the scientists use density function theory calculations and kinetic modeling to map out phase changes under different reaction conditions. With this approach they can discover how reaction conditions affect the active phases of the catalyst and how they can be used to influence catalytic performance.
Newswise — UPTON, N.Y. — Chemists at the U.S. Department of Energy’s (DOE) Brookhaven National Laboratory have developed a new theoretical framework for more accurately predicting the behavior of catalysts. These collections of atoms lower the energy needed for countless chemical reactions. The study reveals how conditions such as temperature and pressure can change a catalyst’s structure, efficiency, and even the products it makes. The findings are published in the journal Chem Catalysis.
“Our results highlight the significant impact the reaction environment can have on catalytic performance,” said Ping Liu, a theorist in Brookhaven Lab’s Chemistry Division who is also an adjunct professor at Stony Brook University (SBU) and oversaw the research. “We show that these catalyst-environment interactions can be used to tune the efficiency and selectivity of catalysts, which could point to new ways to design better catalysts.”
For the study, the scientists modeled catalysts that help hydrogen (H2) convert carbon dioxide (CO2), a greenhouse gas, into a range of other products, including methanol. The catalysts were made of palladium (Pd) partnered with other metals — zinc (Zn) or silver (Ag) — that scientists had previously shown to be effective for the “CO2 hydrogenation” reaction.
They were motivated by a big discrepancy in earlier research on this reaction. In previously published experiments, metallic palladium preferentially produced formic acid (HCOOH). But theoretical calculations predicted that methanol (H3COH) should be the most energetically favorable product.
“This contradiction between theory and experiment made us wonder why there are differences. What are we missing?” Liu asked.
Hong Zhang, Liu’s graduate student at SBU and the first author on the paper, designed a way to find out by modeling what happens during the reaction.
“We developed a framework based on density function theory and kinetic modeling to capture the dynamic behavior and structure of the catalyst under operational reaction conditions,” Zhang said.
The density function theory calculations determine the most likely arrangements and interactions of atoms. The kinetic modeling shows how reactants are transformed from one step of the reaction to the next through a series of intermediates. The idea behind this approach is to bridge the gap between studies of catalysts in their purest, just-made condition and studies that look at catalysts after a reaction is complete.
“The reality is that a catalyst often undergoes significant structural changes or phase transitions in the reaction environment,” Liu said. “But those reaction-driven dynamics are difficult to capture experimentally at the atomic level, even using the amazing characterization tools we have for studying catalysts in real time.”
She noted that the new modeling framework, together with the experimental characterization tools, will provide the precise understanding of catalytic mechanisms that’s needed to guide future catalyst designs.
The modeling framework
Zhang explained how the framework works.
“We start with modeling the as-prepared catalyst — in this case, zinc deposited onto the palladium surface,” he said. “Experimentally, after preparing the catalyst, scientists expose the sample to the mixture of hydrogen and carbon dioxide. We do the same thing using modeling,” Zhang said.
“We consider many representative ‘species’ — reactants and intermediates — that could be present or could form in the particular reaction conditions and have the potential to be stable, as well as how the surface of the catalyst might change.”
Then, the scientists mapped out those “phase changes” in the catalyst under different pressures of carbon dioxide and hydrogen at different temperatures. They discovered which conditions pushed the reaction forward and changed the pathway to produce different products.
At room temperature, they found that the catalytic surface was essentially covered with hydrogen, which blocked the reaction from starting. Raising the temperature produced hydrogen vacancies and gave carbon dioxide access to the active palladium sites. That access initiated the conversion of carbon dioxide and hydrogen to formic acid.
“Going to higher temperatures, we saw even more of a decrease in hydrogen coverage,” Liu said.
More hydrogen vacancies further increased the conversion of carbon dioxide.
“This was expected since most reactions proceed faster at higher temperatures,” Liu explained. “But we also saw a change in selectivity. The reaction went from producing formic acid to producing more and more carbon monoxide and methanol,” she said.
The change in selectivity was somewhat unexpected, but it explained the prior discrepancy between theory and experiment.
“We found that changing the temperature actually changes the active sites of the catalyst, from single hydrogen vacancies to dimers or trimers — pairs or triplets — of missing hydrogens,” Liu said. “Those larger hydrogen vacancies change the catalyst’s selectivity to favor methanol production,” she said.
The scientists then validated and verified that the framework will work for modeling other catalysts. They tested it on pure palladium, a bulk alloy of palladium and zinc where the zinc is not just on the subsurface layer, and a bulk alloy of palladium and silver.
“We simply calculated the surface diagram of these catalysts under the experimental conditions that others have studied. Depending on the coverage of the surface in these conditions, we can easily predict the selectivity,” Liu said. “In all three cases, the framework we developed can accurately describe the experimentally observed selectivity with significantly reduced computing cost.”
This work, she said, has strengthened the interaction of the theoretical and experimental approaches to understanding catalyst structures and mechanisms, as well as how reaction conditions can be used to tune catalytic activity.
“This whole framework goes way beyond the CO2 hydrogenation reaction and the palladium-based catalysts,” Liu said. “It provides a way to gain a deeper understanding of catalysts’ active sites and how they work under operating conditions. This will help establish a structure-catalysis relationship with enhanced precision, which is essential for the design of active and selective catalysts.”
This work was funded by the DOE Office of Science. The calculations used computational resources at the Center for Functional Nanomaterials (CFN), a DOE Office of Science user facility at Brookhaven Lab, and the high-performance SeaWulf computing system of Stony Brook University, made possible by a grant from the National Science Foundation.
Brookhaven National Laboratory is supported by the Office of Science of the U.S. Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, visit science.energy.gov.
Follow @BrookhavenLab on social media. Find us on Instagram, LinkedIn, X, and Facebook.